- Review
- Open access
- Published:
Molecular epidemiology, genetic variability and evolution of HTLV-1 with special emphasis on African genotypes
Retrovirology volume 16, Article number: 39 (2019)
Abstract
Human T cell leukemia virus (HTLV-1) is an oncoretrovirus that infects at least 10 million people worldwide. HTLV-1 exhibits a remarkable genetic stability, however, viral strains have been classified in several genotypes and subgroups, which often mirror the geographic origin of the viral strain. The Cosmopolitan genotype HTLV-1a, can be subdivided into geographically related subgroups, e.g. Transcontinental (a-TC), Japanese (a-Jpn), West-African (a-WA), North-African (a-NA), and Senegalese (a-Sen). Within each subgroup, the genetic diversity is low. Genotype HTLV-1b is found in Central Africa; it is the major genotype in Gabon, Cameroon and Democratic Republic of Congo. While strains from the HTLV-1d genotype represent only a few percent of the strains present in Central African countries, genotypes -e, -f, and -g have been only reported sporadically in particular in Cameroon Gabon, and Central African Republic. HTLV-1c genotype, which is found exclusively in Australo-Melanesia, is the most divergent genotype. This reflects an ancient speciation, with a long period of isolation of the infected populations in the different islands of this region (Australia, Papua New Guinea, Solomon Islands and Vanuatu archipelago). Until now, no viral genotype or subgroup is associated with a specific HTLV-1-associated disease. HTLV-1 originates from a simian reservoir (STLV-1); it derives from interspecies zoonotic transmission from non-human primates to humans (ancient or recent). In this review, we describe the genetic diversity of HTLV-1, and analyze the molecular mechanisms that are at play in HTLV-1 evolution. Similar to other retroviruses, HTLV-1 evolves either through accumulation of point mutations or recombination. Molecular studies point to a fairly low evolution rate of HTLV-1 (between 5.6E−7 and 1.5E−6 substitutions/site/year), supposedly because the virus persists within the host via clonal expansion (instead of new infectious cycles that use reverse transcriptase).
Background
The human T-cell lymphotropic virus (or T-cell leukemia virus) type 1 (HTLV-1), discovered in 1980, has been identified as the first human oncoretrovirus [1]. HTLV-1 is a member of the Retroviridae family, the Orthoretrovirinae subfamily and the Deltaretrovirus genus, which includes bovine leukemia virus (BLV) and T-lymphotropic viruses infecting primates (PTLV). PTLVs consist of simian T-lymphotropic viruses (STLVs) type 1 to 4, which infect non-human primates and human T-lymphotropic viruses type 1–4.
HTLV-1 is the etiological agent of two main very severe diseases: a lympho-proliferative disorder, of mainly CD4 T-cells, named adult T-cell leukemia/lymphoma (ATL) [2], and a chronic neuromyelopathy named tropical spastic paraparesis/HTLV-1 associated myelopathy (TSP/HAM) [3, 4]. HTLV-1 is also associated with other inflammatory diseases including infective dermatitis, some forms of uveitis, myopathies, and bronchiectasis [5].
At least 5 to 10 million people are infected with HTLV-1 worldwide. The known high endemic areas for HTLV-1 are Southwestern Japan, the Caribbean region, parts of South America, sub-Saharan Africa, some foci in the Middle East, and Australo-Melanesia [6,7,8]. The origin of this puzzling geographical (and often ethnic) repartition is likely related to a founder effect in isolated groups where elevated viral transmission rate have persisted. HTLV-1 transmission occurs through sexual intercourse, prolonged breast-feeding, or blood transfusion. Upon leukoreduction, HTLV-1 transmission during transfusion is reduced, evidencing the importance of cell-associated virus in this case [9, 10]. HTLV-1 seroprevalence increases with age, is usually higher in women, and reaches 40% in some highly endemic areas [6,7,8, 11].
HTLV-1 genotypes: classification and geographical distribution
The first HTLV-1 complete sequence (ATK prototype) was obtained in 1983 [12]. It originated from a Japanese patient with ATL. In the following years, many sequences were generated and revealed low genetic variability [13,14,15,16]—when compared to HIV-1 for instance [17]. Interestingly, no evidence for a specific mutation associated with TSP/HAM or ATL was found. In contrast, some nucleotide substitutions observed among HTLV-1 strains were specific to the geographic origin of the patients [18].
Three major molecular genotypes (or subtypes) have been successively identified: the Cosmopolitan a-genotype, the Central African b-genotype, and the Australo-Melanesian c-genotype (Table 1, and Figs. 1 and 2). Other minor genotypes have also been characterized in Central Africa: genotypes -d, -e, -f and -g (Table 1, and Figs. 1, 2, 3) [6, 8]. There is no definite rule for the definition of each genotype, but each genotype is supported by phylogenetic studies (Fig. 3), and intragenotypic variability is lower than intergenotype variability.

Geographical distribution of the seven main molecular genotypes of HTLV-1 (a–g) and major pathways for the spread of the virus through the movements of infected populations

Map of Africa showing the general distribution of HTLV-1 genotypes across the continent. The proportion of the different HTLV-1 genotypes and subgroups is presented for each African country. This figure incorporates the information from papers of molecular epidemiology available on PubMed [20, 21, 23,24,25,26,27, 30, 41, 44,45,46, 55, 68, 135,136,137,138,139,140,141,142,143,144]. It also incorporates results from two manuscripts in preparation (Cassar et al. and Filippone et al.), notably concerning the situation in Benin, Sierra Leone, Western Sahara, and Madagascar, where no data were available to our knowledge. Countries without indications have no informative published data on HTLV-1 genotypes between 1994 and 2019. The size of the circles is proportional to the number of strains identified. The smallest size corresponds to 1 characterized strain, the intermediate sizes to a maximum of 5 or 29 strains and the largest to a minimum of 30 strains. HTLV-1a-North African (HTLV-1 a-NA), HTLV-1a-Senegalese (HTLV-1 a-Sen), HTLV-1a-West African (HTLV-1 a-WA), HTLV-1b and HTLV-1a-Transcontinental (HTLV-1 a-TC) are the most common throughout the continent in North, West, Central and the Austral parts respectively. HTLV-1 d, -e, -f and-g have been identified in Central Africa (Cameroon, Central African Republic, and Gabon)

Phylogenetic representation of the HTLV-1 genotypes and subgroups. An alignment of complete LTR sequences (774-nt long) from 178 HTLV-1 strains was obtained. The unrooted phylogenetic tree was generated with the neighbor-joining method using the GTR model (gamma = 0.4953). Branch lengths are drawn to scale, with the bar indicating 0.01 nucleotide replacement per site. Numbers on each node indicate the percentage of bootstrap samples (of 1000 replicates). HTLV-1 genotypes (a–g) and subgroups (within HTLV-1a and HTLV-1c) are presented. References strains (presented in the table) are indicated in the tree, except Mel1 and Ethio10 for which the complete LTR sequence is not available
The Cosmopolitan a-genotype is the most frequently reported clade and is distributed worldwide. Indeed, it is present in various areas such as Japan, the Caribbean region, Central and South America, West and South Africa, the Middle East, and Europe. This genotype can be further divided into geographically related subgroups. Subgroups are monophyletic clades that can emerge within a genotype, but inter-subgroup genetic diversity is low thus it cannot be considered as a genotype per se. The existence of subgroups suggests that viruses have spread with the migration of ancient infected populations, and have been genetically isolated for centuries or thousands of years.
The initial classification comprised the Transcontinental A subgroup, the Japanese B subgroup, the West-African C subgroup, and the North African D subgroup; they are now referred as a-TC, a-Jpn, a-WA, and a-NA, respectively (Table 1, and Fig. 3) [19,20,21]. More recently, the E/a-Per subgroup, comprising 2 strains from Black Peruvian, was defined [22]; based on partial segment of LTR, a F subgroup has also been identified, especially in an Ethiopian patient [23]. Lastly, we have added in 2006, a Senegalese subgroup (a-Sen), which has also been named “Trans-Saharan” or clade W within the HTLV-1aD subgroup [24,25,26].
-
The transcontinental (TC) subgroup is present on all continents. The overall nucleotide variability within subgroup a-TC is low: it can reach 0–2.5% in the gp21-env gene and 0–2% in the LTR region [27]. It is believed that this low genetic variability reflects the recent dissemination of these strains. In particular, the slave trade from Africa to America, which peaked in the eighteenth century, may represent one of the major paths of recent dissemination [22, 28, 29]. Indeed, HTLV-1 strains found in South Africa, Mozambique, Zimbabwe, Swaziland, and Angola cannot be distinguished from strains found in Brazil [6, 7, 30,31,32]. Additionally, in some studies, clades within the a-TC subgroup have been identified such as South African clusters, Latin-American clusters, and a Middle Eastern cluster [22, 33, 34] (Fig. 4).
Fig. 4 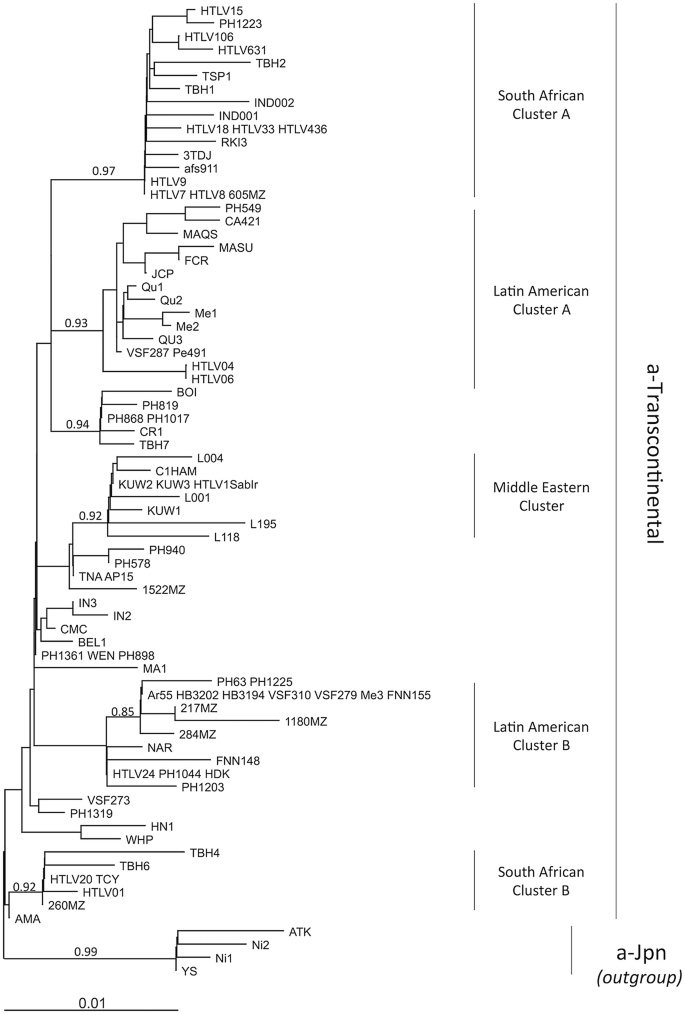
Diverse clusters can be identified within the HTLV-1a-TC subgroup. An alignment of LTR sequences (519-nt long) from 91 HTLV-1a-TC strains was obtained. Sequences from HTLV-1a-Jpn were used as outgroup. The phylogenetic tree was generated with the neighbor-joining method using the GTR model (gamma = 0.4953). Horizontal branch lengths are drawn to scale, with the bar indicating 0.01 nucleotide replacement per site. Values correspond to the approximate likelihood-ratio test for each clade
-
In Japan, strains from the a-TC subgroup coexist with Japanese specific strains [35,36,37,38,39]. The ratio between these two subgroups differs depending on geographical areas and local populations. While the a-TC subgroup is highly predominant among the Ainu in the North and residents of Okinawa (Southwest Japan), the a-Jpn subgroup is predominant among the Wajin population in mainland Japan. Ryukyuans are infected with strains of both subtypes. The reasons for such ethnic and geographical distribution are still under debate. It is believed that the ancestors of the Wajin population were infected when arriving in Japan, and that this virus then evolved into the HTLV-1 a-Jpn. HTLV-1 a-TC may have been introduced more recently in Japan.
-
In Côte d’Ivoire and Ghana, the majority of HTLV-1 strains belong to the West African subgroup (Fig. 2) [40, 41]. a-WA strains were also introduced in South America via the Slave Trade: a-WA strains are found among the Noir-Marron populations living in French Guiana and Surinam [42]. Noir-Marrons are descendants of the slaves who escaped from plantations in the Dutch colony of Surinam during the sixteenth and early seventeenth century. The Noir-Marron have strong genetic affinities close to African populations from the bight of Benin, which is consistent with their predominant HTLV-1 genetic subtype [29, 43].
-
The Senegalese subgroup represents, by definition, the major subgroup present in Senegal (Fig. 2) [25, 26]. It is also present in neighboring countries such as Gambia, Guinea-Bissau, and Mali [24, 44]. In addition, the a-Sen strains are found, but more rarely, in Côte d’Ivoire and Ghana. This is probably a testimony of frequent migrations, some still ongoing, of people from Senegal and neighboring countries to other parts of West Africa.
-
The North African subgroup is mainly present in Algeria, Morocco, Mauritania, Western Sahara, and Mali (Fig. 2) [21, 41]. It can also be found sporadically in other West African countries such as Senegal, Guinea, Côte d’Ivoire, and Ghana.

The Central African b-genotype is most frequently found in Central Africa, i.e. Cameroon, Gabon, CAR, DRC, and Nigeria (Fig. 2). It represents more than 90% of the strains found in Gabon and DRC [27, 45, 46]. HTLV-1b strains differ from HTLV-1a by 2–3% at the nucleotide level (compared to the ATK reference strain) [27]. As for HTLV-1a, strains can cluster according to the geographical origin: HTLV-1 strains from DRC are closer to each other than to strains found in South Cameroon and Gabon, for example [46].
The Australo-Melanesian c-genotype is the most divergent: the genetic nucleotide variability can reach 6–9% when compared to the reference ATK prototype. This reflects an ancient speciation, with a long period of isolation of infected populations living in the different islands of this Pacific region. HTLV-1c was first described in a small group of hunter-horticulturalists living in the fringe highlands of Papua New Guinea (PNG) [47,48,49] and among people of Melanesian origin living in the Solomon Islands [49, 50]. Since, HTLV-1c strains have also been found among residents from Central Australia, the Vanuatu Islands, and New Caledonia [51,52,53]. As with other genotypes, genetic clades that mirror geography can be identified within the HTLV-1c genotype. Phylogenetic analyses indicate the existence of a subgroup composed of strains from the Solomon Islands, the Vanuatu archipelago and New Caledonia (Melanesia subgroup), on the one hand, and an Australian subgroup, on the other (Fig. 3). The Australian subgroup can be further subdivided into two clades (North and South) [53] (Fig. 3).
Other genotypes d, e, f and g have been reported in Central Africa, mainly in Cameroon, Gabon, DRC and CAR [27, 54, 55] (Table 1, and Fig. 2). HTLV-1d can represent up to 3% of the HTLV-1 strains in this region [45]; HTLV-1 e–g strains have been reported sporadically.
The genetic organization differs according to molecular genotypes
HTLV-1 is a complex retrovirus: in addition to structural and regulatory proteins, it encodes several accessory proteins (also called auxiliary proteins). In the HTLV-1a genome, two open reading frames (ORFs) encode four accessory proteins: p12 is encoded by ORFI and can be cleaved into p8; p13 and p30 are encoded by ORFII and are obtained by alternative splicing [56]. These proteins display functions essential for viral persistence in vivo: p12 facilitates immune escape, p8 enables viral propagation, p30 is a negative regulator of viral replication and favors viral persistence, and p13 modulates the cellular response to oxidative stress and allows infected cells to survive [57, 58]. It was early found that deletion of accessory ORFs limits the replication capacity of HTLV-1 in animal models [59], although in some cases mutation in the accessory ORF may have led to disruption of the hbz ORF. Valeri et al. [60] generated a virus deleted for ORF I, with the conservation of hbz. This virus could persist in the rabbit model, but could not persist in Macaques. Thus, the importance of accessory proteins may depend on the host species. Interestingly, some replication may still occur in the latter model as some revertants could appear. Some variability of these accessory proteins has been reported, particularly with regard to p12. Truncated forms of p12 have been described in Japan and South America [61,62,63]. Moreover, two isoforms at position 88 (R/K), which can be linked to different levels of protein expression and degradation, have also been observed [64]. However, it is unclear whether this diversity has an impact on viral expression and pathogenesis in vivo.
The importance of accessory proteins in vivo has been regularly questioned. A HTLV-1a strain deleted for p12 has been described in 3 siblings, suggesting that this virus was transmitted by their mother and is therefore capable of transmission, replication, and persistence in vivo [61]. Similarly, in closely related BLV, the mutation of accessory proteins (R3 and G4) attenuates the virus; the attenuated virus can still replicate and, in the long term, cancers can still appear (although rarely) [65]. Finally, a recent in silico study comparing the complete PTLV-1 genomes available on GenBank confirmed, as expected, that each complete HTLV-1a strain has accessory ORFs and encodes for the 4 proteins. In contrast, strains of the HTLV-1c and -1b subtypes lack some accessory genes [66]. The start codon of ORF I is missing from the complete HTLV-1c and HTLV-1b sequences. Moreover, the splicing acceptor required to generate the mRNA encoding p30 is mutated and may not be functional.
The absence of accessory ORFs, as suggested in the in silico analysis, may indicate that: (1) the encoded proteins are not essential for viral replication in vivo, (2) there are compensatory mutations in the HTLV-1b and HTLV-1c genomes that turn accessory proteins optional, or (3) there are alternative accessory proteins for these viral subtypes. The latter hypothesis is the most likely. Indeed, although the start codon is absent from the ORFII, the ORF contains no additional stop codon. This may suggest a selective pressure to keep the ORF open. The Franchini’s laboratory recently suggested that alternative splicing could lead to the synthesis of p16, an alternative protein to p12 (personal communication). In conclusion, the genetic organization and accessory genes may be different between viral genotypes.
HTLV-1 originates from its simian counterpart through interspecies transmission
Many non-human primates (NHPs) are endemic for STLV-1, the simian counterparts of HTLV-1: STLV-1 can be found in chimpanzees, gorillas, mandrills, baboons, several species of African monkey, a wide range of macaques, and orangutans [67,68,69,70,71,72,73,74]. Clonal proliferation of STLV-1 infected CD4 T-cells has been reported in many NHP species [75]. ATLs have also been reported in a series of STLV-1 infected NHPs [76,77,78].
Interspecies transmission can occur, and is currently ongoing in Central Africa. STLV-1 can be transmitted to humans through infected body fluids, such as saliva and blood. Epidemiological studies have recently found that a severe bite by an ape or a monkey is a major risk factor for HTLV-1 infection in NHP hunters (especially Pygmies) in West Central Africa [79, 80].
It is thus believed that the different HTLV-1 genotypes have originated from ancient interspecies transmission of STLV-1. It is supported by the fact that STLV-1 infecting chimpanzees and gorillas in South Cameroon cannot be distinguished from HTLV-1b strains [80,81,82]. Similarly, STLV-1d is endemic in Mandrills and C. nictitans in Central Africa [67, 70, 73], and STLV-1e and -f are detected in monkeys in Cameroon [67, 83].
However, the case is different for HTLV-1a and -1c. There is no known STLV-1 closely related to these two human genotypes. For HTLV-1a, it can be assumed that either the simian reservoir has not been described yet, or the simian ancestors may have disappeared since the virus was transmitted to humans. For HTLV-1c, the case is even more complex. Indeed, monkeys have never been present in the Australo-Melanesian region. As a result, interspecies transmission of STLV-1 to humans could not occur on these islands. Therefore, it is proposed that HTLV-1c was acquired by proto-Australo-Melanesians during their migration through Southeast Asia, and that populations that reached the highlands of Papua New Guinea were already HTLV-1 infected. The infected populations would then have disseminated, together with their virus, throughout the Australo-Melanesian region [50, 53, 84,85,86,87].
In Asia, STLV-1 is found in many species of macaques [69, 74]. Macaque STLV-1 forms a paraphyletic clade composed of genetically very distant strains [66]. These strains are so distinct that some authors have considered that STLV-1 found in macaca artoides could constitute a novel genotype, called STLV-5 [88]. Intriguingly, zoonotic transmission of STLV-1 has never been reported in Asia, despite a high endemicity of STLV-1 among macaques, and frequent contacts between monkeys and humans in Asia (as evidenced by the transmission of other retroviruses, such as Foamy virus [89, 90]. The reasons for such an apparent restriction of Asian STLV-1 in humans remain unknown. We have recently speculated that STLV-1 from macaques do not express any accessory proteins necessary for viral persistence in the human host [66].
Mechanisms of evolution of HTLV-1
Both recombination and point mutations contribute to the genetic variation of retroviruses. However, until recently, recombination was disregarded when considering HTLV-1 evolution. Indeed, no recombination event had been identified for HTLV-1. The absence of recombination was supported by the fact that no superinfection at the cellular level had been described [91]. Recently, we have identified the first recombinant HTLV-1 strains [41]. We have found that some strains collected from individuals in North Africa (a-NA) are the result of a recombination between HTLV-1 strains related to strains currently present in Senegal (a-Sen) and West Africa (a-WA) (Fig. 3). The recombination site was located at the U3-R junction, suggesting that the recombination event may have occurred during reverse transcription (RT). Ongoing studies have confirmed such findings and identified other recombinants among HTLV-1 strains from West and North Africa. (Cassar et al. in preparation). However, we assume that recombination may be a rare event for HTLV-1, and that the main evolution mechanism for HTLV-1 would be the accumulation of point mutations.
Some intra-individual viral genetic diversity has been reported. Ehrlich et al. [92] found, when studying a 173 bp-long fragment of env, that 16 of the 19 samples displayed genetic variants. Many mutations could be linked to cytidine deaminase activity. Apart from the G>A transition, 7 samples (out of 19) were composed of multiple strains, suggesting the presence of HTLV-1 quasi-species (or multiple infection).
The origin of such diversity is often attributed to the RT. Indeed, the mutation rate of HTLV-1 RT is estimated at 7E−6 mutation/site/replication cycle [93], which is quite comparable to HIV-1 RT. The magnitude of the mutation spectrum in HTLV-1 patients is much lower than what is reported for HIV-1 [94], which is often related to the fact that the virus propagates in vivo mainly by clonal expansion. Indeed, RT is mainly limited to primo-infection in HTLV-1 [95]. Consistently, mutations introduced by cellular polymerase are limited, at least in asymptomatic carriers. Gessain et al. [28] followed infected individuals overtime and found no change in the viral sequences (i.e. 522 nt-long env segment). Of note, the authors had followed only 3 individuals for 6 to 20 months, which explain why no mutation emerged. However, by studying the viral genetic diversity within (and between) infected cellular clones, Mortreux et al. [96] suggested that actually most of the mutations found in the samples were still accumulated during clonal expansion, instead of RT.
In a nutshell, the origin of intra-individual genetic diversity is mostly related to genetic instability and mutations that occur during proliferation of infected cells.
HTLV-1 evolution rate and molecular clock
There are two different methods for estimating the evolution rate of HTLV-1. Such an estimate only takes into account single point mutations, and recombinant strains should be excluded.
On the one hand, the mutation rate can be estimated by studying vertical/intrafamilial transmission chains of the virus. In this context, remarkable genetic stability was observed: first, a study in the DRC (ex-Zaïre) revealed that 10 related individuals carried the same virus, without mutation (in a 755-nt segment of the LTR), although one member was also co-infected with a second strain that differed in one nucleotide [97]. This latter was either the result of a secondary infection, or a mutation that had occurred in that particular individual. A follow-up study, combining this family together with families from South America, found only two mutations in the LTR (756 bp-long) and three mutations in env (522 bp-long) within 16 vertical transmission chains [98]. As a result, mutation rates were estimated at 3.5E−6 and 7.3E−6 substitutions/site/year for LTR and env, respectively. In a similar study in Brazil, the estimation was found surprisingly high (2E−5 substitutions/site/year for LTR), supposedly because it was calculated on the basis of 1 mutation on a single mother–child pair [30]. This value may be largely overestimated. Indeed, in Melanesia, the intra-familial genetic heterogeneity is as low as 0–0.2% over 931 nt [99]. This method focuses mainly on vertical transmission of the virus and generates an estimation of the mutation rate in the short time scale.
On the other hand, the mutation rate can be estimated using phylogeny and an anthropological approach, using a dating anchor point for a given clade. Such analyses are based upon several assumptions: (1) the data set is informative, i.e. the genetic variability is not too high and the phylogenetic signal is not saturated. Salemi et al. [100] found that the data set consisting of each codon of the different canonical genes (i.e. gag, pol, env) were informative for studying all PTLVs (PTLV-1–2 and 3). Similarly, when considering PTLV-1 only, the LTR sequences are also informative [101]. (2) The mutation rate is quite comparable between species (HTLV/STLV) and viral types (PTLV-1/2/3). HTLV and STLV are often considered together in the different analyses. Similarly, PTLV-1 and PTLV-2 are often joined in the studies [100,101,102]. However, it has been shown that HTLV-2 strains isolated from IDUs evolve significantly faster than HTLV-2 strains in an endemic context. Thus HTLV-2 strains from IDUs should be discarded. (3) Either the molecular clock hypothesis is valid or not; in this latter case, a ‘relax clock’ model should be used through Bayesian statistical analysis. The different published papers diverge on this particular point. Salemi et al. [100] found that a data set comprising the 3rd codon of the canonical genes could support the molecular clock hypothesis, when excluding the HTLV-2 IVDU strains. Instead, Lemay et al. [102] preferred studying the 3 codon altogether, and used a Bayesian approach in order to implement a relaxed clock model. When studying HTLV-4, Switzer et al. [88] found saturation on the 3rd codon, and the data set consisting of the 1st and 2nd codon was not suitable with the molecular clock hypothesis. They also had to use a Bayesian approach.
The calibration values for the molecular clock can be major points of debate, and are based on strong assumptions.
The most commonly used date to estimate the time scale for PTLV evolution is the divergence date between HTLV-1c and PTLV1a/b, which is estimated between 40,000 and 60,000 years ago [88, 100,101,102]. It was at this time that the first populations migrated from Asia to Melanesia. As discussed above, since no simians have ever been detected in Oceania, populations that transmitted HTLV-1 to Australo-Melanesia are considered to have acquired the virus from Indonesian NHPs on their migration route [84]. However, recently, Reid et al. [103] have challenged this dating. They believe HTLV-1 was introduced into Australo-Melanesia much recently, during a more massive wave of migration that originates from India, about 4000 years ago. This change in dating would results in a different and much higher mutation rate.
Another possible date is the divergence between HTLV-2a and -2c (in studies combining the two types of viruses). Indeed, these two clades are composed exclusively of strains present in Amerindian populations. It was therefore proposed that they share a common ancestor who reached the Americas at the time of human migration on the Bering Strait. Thus, the HTLV-2a/c node is dated at 25,000 ± 5,000 years ago [100, 104].
In conclusion, depending on the different models and assumptions, the estimated mutation rates vary from 5.6E−7 [102] to 1.5E−6 [101] and 6.2E−6 [103] subst/site/year, for the LTR. When considering coding regions, the substitution rate is between 2.1E−7 and 8E−7 subst/site/year (assuming a Bayesian relaxed molecular clock) [88, 102].
Conclusions: major unanswered questions concerning HTLV-1 molecular variety
Despite a good understanding of the genetic diversity and evolution mechanisms of HTLV-1, many questions remain concerning the origins of some groups infected with HTLV-1, and the pathogenicity of each genotype.
-
1.
Several European countries (e.g. France, Great Britain and Spain) regularly report cases of HTLV-1 infection (among blood donors or pregnant women) or HTLV-1 associated diseases [105,106,107]. In these countries, most of the infected individuals come from regions where HTLV-1 is highly endemic, such as the Caribbean area, sub-Saharan Africa, and South America. In contrast, Romania has a high prevalence of HTLV-1 infection [108, 109], but there is no evidence of significant migrations from HTLV-1 endemicity areas. Thus, Romania seems to be a nucleus of endogenous endemicity in Europe. The origins of HTLV-1 in Romania are unknown. From a molecular point of view, the viral strains present in Romania belong to the TC subgroup of the Cosmopolitan a-genotype [110, 111]. Extensive epidemiological and molecular studies are being conducted in order to get new insights into the origin and dissemination of HTLV-1 infection in Romania.
-
2.
HTLV-1 has been found in many native populations in the Americas, such as the Inuit in Canada and the USA, the Quetchua in Peru, the Mapuche in Chile, and indigenous groups from Argentina [112,113,114,115]. Most strains belong to the large a-TC subgroup; in some cases, geographical clusters can be identified (small and large Latin American Clusters, Jujuy specific cluster, etc.) [112,113,114,115,116]. The origin of such infection is still controversial: either the virus has recently been acquired—through contacts with infected individuals from Africa, following the slave trade for example [30,31,32, 117]—or the virus was introduced during the initial settlement of the American continent, with the migration of infected populations through the Behring Strait [118,119,120].
-
3.
The modes of dissemination of HTLV-1 in the Middle East and Asia remain to be clarified. Regions of the Middle East (e.g. areas of Iran and Kuwait) have been found endemic for HTLV-1 [121, 122]. A few strains have been characterized, and suggest that there is a Middle Eastern cluster within the HTLV-1a TC subgroup [33, 34, 123]. Interestingly, some strains found in India are closely related to strains from the Middle East [124]. Thus, infected populations have migrated between these regions. Some suggest that the ancient Silk Road, which linked China to Antioch (now in Turkey), could also have been a Road for the dissemination of HTLV-1.
-
4.
The importance of human migrations in the modern area will likely modify the distribution of HTLV-1 and lead to a mixing of genotypes and subtypes. Indeed, the Tokyo metropolitan area may become a hotspot of endemicity for HTLV-1 as individuals migrate from endemic areas such as the Kyushu-Okinawa region [125]. In some cases, long-distance migrations occur and lead to a wider distribution of a previously geographically restricted subtype. Thus, a-Jpn strains have been found in other countries, such as Peru [22], Hawaii USA [126], and South Africa [127] (Fig. 2).
-
5.
There is no clear evidence of specific mutations in the HTLV-1 genome that would render the virus more pathogenic [128, 129]. However, most of the reported cases of ATL and TSP/HAM correspond to individuals infected with HTLV-1 strains from the a-genotype. Does this mean that this genotype is more pathogenic than the others? For instance, it has been suggested that Australian HTLV-1c strains might be less oncogenic, more likely to induce inflammatory diseases (such as bronchiectasis) than tumors [130,131,132]. Since, ATL cases have been reported in HTLV-1c carriers [133, 134]. One of the reasons why the proportion of ATL appears to be lower among Indigenous Australians may be related to the fact that this population is younger and has a shorter life expectancy; it may also be underreported. In order to clearly answer this particular point, cohort-based prospective studies on HTLV-1b and HTLV-1c populations are needed.
Availability of data and materials
Not applicable.
References
Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA. 1980;77:7415–9.
Takatsuki K, Uchiyama T, Sagawa K, Yodoi J. Adult T cell leukemia in Japan. In: Seno S, Takaku F, Irino S, editors. Topics in hematology. Amsterdam: Excerpta Medica; 1977. p. 73–7.
Gessain A, Barin F, Vernant JC, Gout O, Maurs L, Calender A, de The G. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet. 1985;2:407–10.
Osame M, Usuku K, Izumo S, Ijichi N, Amitani H, Igata A, Matsumoto M, Tara M. HTLV-I associated myelopathy, a new clinical entity. Lancet. 1986;1:1031–2.
Martin F, Taylor GP, Jacobson S. Inflammatory manifestations of HTLV-1 and their therapeutic options. Expert Rev Clin Immunol. 2014;10:1531–46.
Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol. 2012;3:388.
Proietti FA, Carneiro-Proietti AB, Catalan-Soares BC, Murphy EL. Global epidemiology of HTLV-I infection and associated diseases. Oncogene. 2005;24:6058–68.
Verdonck K, Gonzalez E, Van Dooren S, Vandamme AM, Vanham G, Gotuzzo E. Human T-lymphotropic virus 1: recent knowledge about an ancient infection. Lancet Infect Dis. 2007;7:266–81.
Hewitt PE, Davison K, Howell DR, Taylor GP. Human T-lymphotropic virus lookback in NHS blood and transplant (England) reveals the efficacy of leukoreduction. Transfusion. 2013;53:2168–75.
Cesaire R, Kerob-Bauchet B, Bourdonne O, Maier H, Amar KO, Halbout P, Dehee A, Desire N, Dantin F, Bera O, Lezin A. Evaluation of HTLV-I removal by filtration of blood cell components in a routine setting. Transfusion. 2004;44:42–8.
Murphy EL, Figueroa JP, Gibbs WN, Holding-Cobham M, Cranston B, Malley K, Bodner AJ, Alexander SS, Blattner WA. Human T-lymphotropic virus type I (HTLV-I) seroprevalence in Jamaica. I. Demographic determinants. Am J Epidemiol. 1991;133:1114–24.
Seiki M, Hattori S, Hirayama Y, Yoshida M. Human adult T-cell leukemia virus: complete nucleotide sequence of the provirus genome integrated in leukemia cell DNA. Proc Natl Acad Sci USA. 1983;80:3618–22.
Bazarbachi A, Huang M, Gessain A, Saal F, Saib A, Peries J, De The H, Galibert F. Human T-cell-leukemia virus type I in post-transfusional spastic paraparesis: complete proviral sequence from uncultured blood cells. Int J Cancer. 1995;63:494–9.
Malik KT, Even J, Karpas A. Molecular cloning and complete nucleotide sequence of an adult T cell leukaemia virus/human T cell leukaemia virus type I (ATLV/HTLV-I) isolate of Caribbean origin: relationship to other members of the ATLV/HTLV-I subgroup. J Gen Virol. 1988;69(Pt 7):1695–710.
Paine E, Garcia J, Philpott TC, Shaw G, Ratner L. Limited sequence variation in human T-lymphotropic virus type 1 isolates from North American and African patients. Virology. 1991;182:111–23.
Evangelista A, Maroushek S, Minnigan H, Larson A, Retzel E, Haase A, Gonzalez-Dunia D, McFarlin D, Mingioli E, Jacobson S, et al. Nucleotide sequence analysis of a provirus derived from an individual with tropical spastic paraparesis. Microb Pathog. 1990;8:259–78.
Hemelaar J. Implications of HIV diversity for the HIV-1 pandemic. J Infect. 2013;66:391–400.
Komurian F, Pelloquin F, de The G. In vivo genomic variability of human T-cell leukemia virus type I depends more upon geography than upon pathologies. J Virol. 1991;65:3770–8.
Miura T, Fukunaga T, Igarashi T, Yamashita M, Ido E, Funahashi S, Ishida T, Washio K, Ueda S, Hashimoto K, et al. Phylogenetic subtypes of human T-lymphotropic virus type I and their relations to the anthropological background. Proc Natl Acad Sci USA. 1994;91:1124–7.
Vidal AU, Gessain A, Yoshida M, Tekaia F, Garin B, Guillemain B, Schulz T, Farid R, De The G. Phylogenetic classification of human T cell leukaemia/lymphoma virus type I genotypes in five major molecular and geographical subtypes. J Gen Virol. 1994;75(Pt 12):3655–66.
Gasmi M, Farouqi B, d’Incan M, Desgranges C. Long terminal repeat sequence analysis of HTLV type I molecular variants identified in four north African patients. AIDS Res Hum Retrovir. 1994;10:1313–5.
Van Dooren S, Gotuzzo E, Salemi M, Watts D, Audenaert E, Duwe S, Ellerbrok H, Grassmann R, Hagelberg E, Desmyter J, Vandamme AM. Evidence for a post-Columbian introduction of human T-cell lymphotropic virus [type I] [corrected] in Latin America. J Gen Virol. 1998;79(Pt 11):2695–708.
Trevino A, Alcantara LC Jr, Benito R, Caballero E, Aguilera A, Ramos JM, de Mendoza C, Rodriguez C, Garcia J, Rodriguez-Iglesias M, et al. Molecular epidemiology and clinical features of human T cell lymphotropic virus type 1 infection in Spain. AIDS Res Hum Retrovir. 2014;30(9):856–62.
Zehender G, Ebranati E, De Maddalena C, Gianelli E, Riva A, Rusconi S, Massetto B, Rankin F, Acurie M, Galli M. Description of a “trans-Saharan” strain of human T-lymphotropic virus type 1 in West Africa. J Acquir Immune Defic Syndr. 2008;47:269–73.
Mahe A, Meertens L, Ly F, Sow PS, Diop CT, Samb ND, Diop OM, Valensi F, Gessain A. Human T-cell leukaemia/lymphoma virus type 1-associated infective dermatitis in Africa: a report of five cases from Senegal. Br J Dermatol. 2004;150:958–65.
Diop S, Calattini S, Abah-Dakou J, Thiam D, Diakhate L, Gessain A. Seroprevalence and molecular epidemiology of human T-Cell leukemia virus type 1 (HTLV-1) and HTLV-2 in blood donors from Dakar, Senegal. J Clin Microbiol. 2006;44:1550–4.
Mahieux R, Ibrahim F, Mauclere P, Herve V, Michel P, Tekaia F, Chappey C, Garin B, Van Der Ryst E, Guillemain B, et al. Molecular epidemiology of 58 new African human T-cell leukemia virus type 1 (HTLV-1) strains: identification of a new and distinct HTLV-1 molecular subtype in Central Africa and in Pygmies. J Virol. 1997;71:1317–33.
Gessain A, Gallo RC, Franchini G. Low degree of human T-cell leukemia/lymphoma virus type I genetic drift in vivo as a means of monitoring viral transmission and movement of ancient human populations. J Virol. 1992;66:2288–95.
Brucato N, Cassar O, Tonasso L, Tortevoye P, Migot-Nabias F, Plancoulaine S, Guitard E, Larrouy G, Gessain A, Dugoujon JM. The imprint of the slave trade in an African American population: mitochondrial DNA, Y chromosome and HTLV-1 analysis in the Noir Marron of French Guiana. BMC Evol Biol. 2010;10:314.
Alcantara LC, de Oliveira T, Gordon M, Pybus O, Mascarenhas RE, Seixas MO, Goncalves M, Hlela C, Cassol S, Galvao-Castro B. Tracing the origin of Brazilian HTLV-1 as determined by analysis of host and viral genes. AIDS. 2006;20:780–2.
Rego FF, Alcantara LC, Moura Neto JP, Miranda AC, Pereira Ode S, Goncalves Mde S, Galvao-Castro B. HTLV type 1 molecular study in Brazilian villages with African characteristics giving support to the post-Columbian introduction hypothesis. AIDS Res Hum Retrovir. 2008;24:673–7.
Amoussa AE, Wilkinson E, Giovanetti M, de Almeida Rego FF, Araujo TH, de Souza Goncalves M, de Oliveira T, Alcantara LC. HTLV-1aA introduction into Brazil and its association with the trans-Atlantic slave trade. Infect Genet Evol. 2017;48:95–101.
Pirayeshfard L, Sharifi Z, Amini-Kafiabad S, Haghnazari Sadaghiani N. Phylogenetic analysis of HTLV-1 in Iranian blood donors, HIV-1 positive patients and patients with beta thalassemia. J Med Virol. 2018;90:1398–405.
Mirhosseini A, Mohareri M, Arab R, Rezaee SA, Shirdel A, Koshyar MM, Allahyari A, Bari A, Rahimi H, Mozaheb Z, et al. Complete sequence of human T cell leukemia virus type 1 in ATLL patients from Northeast Iran, Mashhad revealed a prematurely terminated protease and an elongated pX open reading frame III. Infect Genet Evol. 2019;73:460–9.
Otani M, Honda N, Xia PC, Eguchi K, Ichikawa T, Watanabe T, Yamaguchi K, Nakao K, Yamamoto T. Distribution of two subgroups of human T-lymphotropic virus type 1 (HTLV-1) in endemic Japan. Trop Med Health. 2012;40:55–8.
Eguchi K, Fujii H, Oshima K, Otani M, Matsuo T, Yamamoto T. Human T-lymphotropic virus type 1 (HTLV-1) genetic typing in Kakeroma Island, an island at the crossroads of the ryukyuans and Wajin in Japan, providing further insights into the origin of the virus in Japan. J Med Virol. 2009;81:1450–6.
Otani M, Eguchi K, Ichikawa T, Takenaka Takano K, Watanabe T, Yamaguchi K, Nakao K, Yamamoto T. Phylogeography of human T-lymphotropic virus type 1 (HTLV-1) lineages endemic to Japan. Trop Med Health. 2012;40:117–24.
Vidal AU, Gessain A, Yoshida M, Mahieux R, Nishioka K, Tekaia F, Rosen L, De The G. Molecular epidemiology of HTLV type I in Japan: evidence for two distinct ancestral lineages with a particular geographical distribution. AIDS Res Hum Retrovir. 1994;10:1557–66.
Yamashita M, Ishida T, Ohkura S, Miura T, Hayami M. Phylogenetic characterization of a new HTLV type 1 from the Ainu in Japan. AIDS Res Hum Retrovir. 2001;17:783–7.
Calvignac-Spencer S, Adjogoua EV, Akoua-Koffi C, Hedemann C, Schubert G, Ellerbrok H, Leendertz SA, Pauli G, Leendertz FH. Origin of human T-lymphotropic virus type 1 in rural Cote d’Ivoire. Emerg Infect Dis. 2012;18:830–3.
Desrames A, Cassar O, Gout O, Hermine O, Taylor GP, Afonso PV, Gessain A. Northern african strains of human T-lymphotropic virus type 1 arose from a recombination event. J Virol. 2014;88:9782–8.
Talarmin A, Vion B, Ureta-Vidal A, Du Fou G, Marty C, Kazanji M. First seroepidemiological study and phylogenetic characterization of human T-cell lymphotropic virus type I and II infection among Amerindians in French Guiana. J Gen Virol. 1999;80(Pt 12):3083–8.
Fortes-Lima C, Gessain A, Ruiz-Linares A, Bortolini MC, Migot-Nabias F, Bellis G, Moreno-Mayar JV, Restrepo BN, Rojas W, Avendano-Tamayo E, et al. Genome-wide ancestry and demographic history of African-descendant maroon communities from French Guiana and Suriname. Am J Hum Genet. 2017;101:725–36.
van Tienen C, de Silva TI, Alcantara LC, Onyango CO, Jarju S, Goncalves N, Vincent T, Aaby P, Whittle H, van der Loeff MS, Cotten M. Molecular epidemiology of endemic human T-lymphotropic virus type 1 in a rural community in Guinea-Bissau. PLoS Negl Trop Dis. 2012;6:e1690.
Caron M, Besson G, Padilla C, Makuwa M, Nkoghe D, Leroy E, Kazanji M. Revisiting human T-cell lymphotropic virus types 1 and 2 infections among rural population in Gabon, central Africa thirty years after the first analysis. PLoS Negl Trop Dis. 2018;12:e0006833.
Hogan CA, Iles J, Frost EH, Giroux G, Cassar O, Gessain A, Dion MJ, Ilunga V, Rambaut A, Yengo-Ki-Ngimbi AE, et al. Epidemic history and iatrogenic transmission of blood-borne viruses in mid-20th century Kinshasa. J Infect Dis. 2016;214:353–60.
Sherman MP, Saksena NK, Dube DK, Yanagihara R, Poiesz BJ. Evolutionary insights on the origin of human T-cell lymphoma/leukemia virus type I (HTLV-I) derived from sequence analysis of a new HTLV-I variant from Papua New Guinea. J Virol. 1992;66:2556–63.
Saksena NK, Sherman MP, Yanagihara R, Dube DK, Poiesz BJ. LTR sequence and phylogenetic analyses of a newly discovered variant of HTLV-I isolated from the Hagahai of Papua New Guinea. Virology. 1992;189:1–9.
Gessain A, Yanagihara R, Franchini G, Garruto RM, Jenkins CL, Ajdukiewicz AB, Gallo RC, Gajdusek DC. Highly divergent molecular variants of human T-lymphotropic virus type I from isolated populations in Papua New Guinea and the Solomon Islands. Proc Natl Acad Sci USA. 1991;88:7694–8.
Gessain A, Boeri E, Yanagihara R, Gallo RC, Franchini G. Complete nucleotide sequence of a highly divergent human T-cell leukemia (lymphotropic) virus type I (HTLV-I) variant from melanesia: genetic and phylogenetic relationship to HTLV-I strains from other geographical regions. J Virol. 1993;67:1015–23.
Cassar O, Capuano C, Bassot S, Charavay F, Duprez R, Afonso PV, Abel M, Walter H, Mera W, Martin PM, et al. Human T lymphotropic virus type 1 subtype C melanesian genetic variants of the Vanuatu Archipelago and Solomon Islands share a common ancestor. J Infect Dis. 2007;196:510–21.
Cassar O, Charavay F, Touzain F, Jeannin P, Grangeon JP, Laumond S, Chungue E, Martin PM, Gessain A. A novel human T-lymphotropic virus type 1c molecular variant in an indigenous individual from New Caledonia, Melanesia. PLoS Negl Trop Dis. 2017;11:e0005278.
Cassar O, Einsiedel L, Afonso PV, Gessain A. Human T-cell lymphotropic virus type 1 subtype C molecular variants among Indigenous Australians: new insights into the molecular epidemiology of HTLV-1 in Australo-Melanesia. PLoS Negl Trop Dis. 2013;7(9):e2418.
Salemi M, Van Dooren S, Audenaert E, Delaporte E, Goubau P, Desmyter J, Vandamme AM. Two new human T-lymphotropic virus type I phylogenetic subtypes in seroindeterminates, a Mbuti pygmy and a Gabonese, have closest relatives among African STLV-I strains. Virology. 1998;246:277–87.
Wolfe ND, Heneine W, Carr JK, Garcia AD, Shanmugam V, Tamoufe U, Torimiro JN, Prosser AT, Lebreton M, Mpoudi-Ngole E, et al. Emergence of unique primate T-lymphotropic viruses among central African bushmeat hunters. Proc Natl Acad Sci USA. 2005;102:7994–9.
Koralnik IJ, Gessain A, Klotman ME, Lo Monico A, Berneman ZN, Franchini G. Protein isoforms encoded by the pX region of human T-cell leukemia/lymphotropic virus type I. Proc Natl Acad Sci USA. 1992;89:8813–7.
Bai XT, Nicot C. Overview on HTLV-1 p12, p8, p30, p13: accomplices in persistent infection and viral pathogenesis. Front Microbiol. 2012;3:400.
Edwards D, Fenizia C, Gold H, de Castro-Amarante MF, Buchmann C, Pise-Masison CA, Franchini G. Orf-I and orf-II-encoded proteins in HTLV-1 infection and persistence. Viruses. 2011;3:861–85.
Lairmore MD, Albrecht B, D’Souza C, Nisbet JW, Ding W, Bartoe JT, Green PL, Zhang W. In vitro and in vivo functional analysis of human T cell lymphotropic virus type 1 pX open reading frames I and II. AIDS Res Hum Retrovir. 2000;16:1757–64.
Valeri VW, Hryniewicz A, Andresen V, Jones K, Fenizia C, Bialuk I, Chung HK, Fukumoto R, Parks RW, Ferrari MG, et al. Requirement of the human T-cell leukemia virus p12 and p30 products for infectivity of human dendritic cells and macaques but not rabbits. Blood. 2010;116:3809–17.
Furukawa Y, Usuku K, Izumo S, Osame M. Human T cell lymphotropic virus type I (HTLV-I) p12I is dispensable for HTLV-I transmission and maintenance of infection in vivo. AIDS Res Hum Retrovir. 2004;20:1092–9.
Iniguez AM, Gastaldello R, Gallego S, Otsuki K, Vicente AC. HTLV-1 p12I protein sequences from South America: truncated proteins and common genetic signatures. AIDS Res Hum Retrovir. 2006;22:466–9.
Rosadas C, Vicente ACP, Zanella L, Cabral-Castro MJ, Peralta JM, Puccioni-Sohler M. First report of HTLV-1 truncated p12 protein in Brazil. Virulence. 2017;8:1445–9.
Martins ML, Soares BC, Ribas JG, Thorun GW, Johnson J, Kroon EG, Carneiro-Prioetti AB, Bonjardim CA. Giph: frequency of p12K and p12R alleles of HTLV type 1 in HAM/TSP patients and in asymptomatic HTLV type 1 carriers. AIDS Res Hum Retrovir. 2002;18:899–902.
Florins A, Gillet N, Boxus M, Kerkhofs P, Kettmann R, Willems L. Even attenuated bovine leukemia virus proviruses can be pathogenic in sheep. J Virol. 2007;81:10195–200.
Afonso PV, Fagrouch Z, Deijs M, Niphuis H, Bogers W, Gessain A, van der Hoek L, Verschoor EJ. Absence of accessory genes in a divergent simian T-lymphotropic virus type 1 isolated from a bonnet macaque (Macaca radiata). PLoS Negl Trop Dis. 2019;13:e0007521.
Locatelli S, Peeters M. Cross-species transmission of simian retroviruses: how and why they could lead to the emergence of new diseases in the human population. Aids. 2012;26:659–73.
Mossoun A, Calvignac-Spencer S, Anoh AE, Pauly MS, Driscoll DA, Michel AO, Nazaire LG, Pfister S, Sabwe P, Thiesen U, et al. Bushmeat hunting and zoonotic transmission of simian T-lymphotropic virus 1 in Tropical West and Central Africa. J Virol. 2017;91:e02479-16.
Ayouba A, Duval L, Liegeois F, Ngin S, Ahuka-Mundeke S, Switzer WM, Delaporte E, Ariey F, Peeters M, Nerrienet E. Nonhuman primate retroviruses from Cambodia: high simian foamy virus prevalence, identification of divergent STLV-1 strains and no evidence of SIV infection. Infect Genet Evol. 2013;18:325–34.
Liegeois F, Lafay B, Switzer WM, Locatelli S, Mpoudi-Ngole E, Loul S, Heneine W, Delaporte E, Peeters M. Identification and molecular characterization of new STLV-1 and STLV-3 strains in wild-caught nonhuman primates in Cameroon. Virology. 2008;371:405–17.
Van Dooren S, Verschoor EJ, Fagrouch Z, Vandamme AM. Phylogeny of primate T lymphotropic virus type 1 (PTLV-1) including various new Asian and African non-human primate strains. Infect Genet Evol. 2007;7:374–81.
Mahieux R, Chappey C, Meertens L, Mauclere P, Lewis J, Gessain A. Molecular characterization and phylogenetic analyses of a new simian T cell lymphotropic virus type 1 in a wild-caught african baboon (Papio anubis) with an indeterminate STLV type 2-like serology. AIDS Res Hum Retrovir. 2000;16:2043–8.
Mahieux R, Chappey C, Georges-Courbot MC, Dubreuil G, Mauclere P, Georges A, Gessain A. Simian T-cell lymphotropic virus type 1 from Mandrillus sphinx as a simian counterpart of human T-cell lymphotropic virus type 1 subtype D. J Virol. 1998;72:10316–22.
Mahieux R, Pecon-Slattery J, Gessain A. Molecular characterization and phylogenetic analyses of a new, highly divergent simian T-cell lymphotropic virus type 1 (STLV-1marc1) in Macaca arctoides. J Virol. 1997;71:6253–8.
Gabet AS, Gessain A, Wattel E. High simian T-cell leukemia virus type 1 proviral loads combined with genetic stability as a result of cell-associated provirus replication in naturally infected, asymptomatic monkeys. Int J Cancer. 2003;107:74–83.
Homma T, Kanki PJ, King NW Jr, Hunt RD, O’Connell MJ, Letvin NL, Daniel MD, Desrosiers RC, Yang CS, Essex M. Lymphoma in macaques: association with virus of human T lymphotrophic family. Science. 1984;225:716–8.
Miura M, Yasunaga J, Tanabe J, Sugata K, Zhao T, Ma G, Miyazato P, Ohshima K, Kaneko A, Watanabe A, et al. Characterization of simian T-cell leukemia virus type 1 in naturally infected Japanese macaques as a model of HTLV-1 infection. Retrovirology. 2013;10:118.
Turpin J, Alais S, Marcais A, Bruneau J, Melamed A, Gadot N, Tanaka Y, Hermine O, Melot S, Lacoste R, et al. Whole body clonality analysis in an aggressive STLV-1 associated leukemia (ATLL) reveals an unexpected clonal complexity. Cancer Lett. 2017;389:78–85.
Kazanji M, Mouinga-Ondeme A, Lekana-Douki-Etenna S, Caron M, Makuwa M, Mahieux R, Gessain A. Origin of HTLV-1 in hunters of nonhuman primates in Central Africa. J Infect Dis. 2015;211:361–5.
Filippone C, Betsem E, Tortevoye P, Cassar O, Bassot S, Froment A, Fontanet A, Gessain A. A severe bite from a non-human primate is a major risk factor for HTLV-1 infection in hunters from Central Africa. Clin Infect Dis. 2015;60(11):1667–76.
Ayouba A, Michem A, Peeters M, Vercammen F. Full-genome characterization of simian T-cell leukemia virus type 1 subtype b from a wild-born captive Gorilla gorilla gorilla with T-cell lymphoma. Genome Announc. 2017;5:e01117-17.
Koralnik IJ, Boeri E, Saxinger WC, Monico AL, Fullen J, Gessain A, Guo HG, Gallo RC, Markham P, Kalyanaraman V, et al. Phylogenetic associations of human and simian T-cell leukemia/lymphotropic virus type I strains: evidence for interspecies transmission. J Virol. 1994;68:2693–707.
Nerrienet E, Meertens L, Kfutwah A, Foupouapouognigni Y, Gessain A. Molecular epidemiology of simian T-lymphotropic virus (STLV) in wild-caught monkeys and apes from Cameroon: a new STLV-1, related to human T-lymphotropic virus subtype F, in a Cercocebus agilis. J Gen Virol. 2001;82:2973–7.
Ibrahim F, de The G, Gessain A. Isolation and characterization of a new simian T-cell leukemia virus type 1 from naturally infected celebes macaques (Macaca tonkeana): complete nucleotide sequence and phylogenetic relationship with the Australo-Melanesian human T-cell leukemia virus type 1. J Virol. 1995;69:6980–93.
de The G. Microbial genomes to write our history. J Infect Dis. 2007;196:499–501.
Yanagihara R. Geographic-specific genotypes or topotypes of human T-cell lymphotropic virus type I as markers for early and recent migrations of human populations. Adv Virus Res. 1994;43:147–86.
Yanagihara R, Saitou N, Nerurkar VR, Song KJ, Bastian I, Franchini G, Gajdusek DC. Molecular phylogeny and dissemination of human T-cell lymphotropic virus type I viewed within the context of primate evolution and human migration. Cell Mol Biol. 1995;41(Suppl 1):S145–61.
Switzer WM, Salemi M, Qari SH, Jia H, Gray RR, Katzourakis A, Marriott SJ, Pryor KN, Wolfe ND, Burke DS, et al. Ancient, independent evolution and distinct molecular features of the novel human T-lymphotropic virus type 4. Retrovirology. 2009;6:9.
Jones-Engel L, Engel GA, Schillaci MA, Rompis A, Putra A, Suaryana KG, Fuentes A, Beer B, Hicks S, White R, et al. Primate-to-human retroviral transmission in Asia. Emerg Infect Dis. 2005;11:1028–35.
Engel GA, Small CT, Soliven K, Feeroz MM, Wang X. Zoonotic simian foamy virus in Bangladesh reflects diverse patterns of transmission and co-infection. Emerg Microbes Infect. 2013;2:1–10.
Cook LB, Rowan AG, Melamed A, Taylor GP, Bangham CR. HTLV-1-infected T cells contain a single integrated provirus in natural infection. Blood. 2012;120:3488–90.
Ehrlich GD, Andrews J, Sherman MP, Greenberg SJ, Poiesz BJ. DNA sequence analysis of the gene encoding the HTLV-I p21e transmembrane protein reveals inter- and intraisolate genetic heterogeneity. Virology. 1992;186:619–27.
Mansky LM. In vivo analysis of human T-cell leukemia virus type 1 reverse transcription accuracy. J Virol. 2000;74:9525–31.
Eberle J, Gurtler L. HIV types, groups, subtypes and recombinant forms: errors in replication, selection pressure and quasispecies. Intervirology. 2012;55:79–83.
Cook LB, Melamed A, Demontis MA, Laydon DJ, Fox JM, Tosswill JH, de Freitas D, Price AD, Medcalf JF, Martin F, et al. Rapid dissemination of human T-lymphotropic virus type 1 during primary infection in transplant recipients. Retrovirology. 2016;13:3.
Mortreux F, Leclercq I, Gabet AS, Leroy A, Westhof E, Gessain A, Wain-Hobson S, Wattel E. Somatic mutation in human T-cell leukemia virus type 1 provirus and flanking cellular sequences during clonal expansion in vivo. J Natl Cancer Inst. 2001;93:367–77.
Liu HF, Vandamme AM, Kazadi K, Carton H, Desmyter J, Goubau P. Familial transmission and minimal sequence variability of human T-lymphotropic virus type I (HTLV-I) in Zaire. AIDS Res Hum Retrovir. 1994;10:1135–42.
Van Dooren S, Pybus OG, Salemi M, Liu HF, Goubau P, Remondegui C, Talarmin A, Gotuzzo E, Alcantara LC, Galvao-Castro B, Vandamme AM. The low evolutionary rate of human T-cell lymphotropic virus type-1 confirmed by analysis of vertical transmission chains. Mol Biol Evol. 2004;21:603–11.
Nerurkar VR, Song KJ, Saitou N, Melland RR, Yanagihara R. Interfamilial and intrafamilial genomic diversity and molecular phylogeny of human T-cell lymphotropic virus type I from Papua New Guinea and the Solomon Islands. Virology. 1993;196:506–13.
Salemi M, Desmyter J, Vandamme AM. Tempo and mode of human and simian T-lymphotropic virus (HTLV/STLV) evolution revealed by analyses of full-genome sequences. Mol Biol Evol. 2000;17:374–86.
Van Dooren S, Salemi M, Vandamme AM. Dating the origin of the African human T-cell lymphotropic virus type-i (HTLV-I) subtypes. Mol Biol Evol. 2001;18:661–71.
Lemey P, Pybus OG, Van Dooren S, Vandamme AM. A Bayesian statistical analysis of human T-cell lymphotropic virus evolutionary rates. Infect Genet Evol. 2005;5:291–8.
Reid MJ, Switzer WM, Schillaci MA, Ragonnet-Cronin M, Joanisse I, Caminiti K, Lowenberger CA, Galdikas BM, Sandstrom PA, Brooks JI. Detailed phylogenetic analysis of primate T-lymphotropic virus type 1 (PTLV-1) sequences from orangutans (Pongo pygmaeus) reveals new insights into the evolutionary history of PTLV-1 in Asia. Infect Genet Evol. 2016;43:434–50.
Mauclère P, Afonso P, Meertens L, Plancoulaine S, Calattini S, Froment A, Van Beveren M, de Thé G, Quintana-Murci L, Mahieux R, Gessain A. HTLV-2B strains, similar to those found in several Amerindians tribes, are endemic in Centra African Bakola Pygmies. J Infect Dis. 2011;203:1316–23.
de Mendoza C, Caballero E, Aguilera A, Requena S, de Lejarazu RO, Piron M, Gonzalez R, Jimenez A, Roc L, Trevino A, et al. Human T-lymphotropic virus type 1 infection and disease in Spain. AIDS. 2017;31:1653–63.
Ireland G, Croxford S, Tosswill J, Raghu R, Davison K, Hewitt P, Simmons R, Taylor G. Human T-lymphotropic viruses (HTLV) in England and Wales, to 2013: testing and diagnoses. Euro Surveill. 2004;2017:22.
Laperche S, Worms B, Pillonel J. Blood safety strategies for human T-cell lymphotropic virus in Europe. Vox Sang. 2009;96:104–10.
Fett NM, Siddiqui J, Creswell CH, Zhang D, Lloyd R, Wood GS. Adult T-cell leukemia/lymphoma in a patient from Romania: a case report and review of the literature. J Cutan Pathol. 2008;35(Suppl 1):32–7.
Paun L, Ispas O, Del Mistro A, Chieco-Bianchi L. HTLV-I in Romania. Eur J Haematol. 1994;52:117–8.
Ellerbrok H, Fleischer C, Salemi M, Reinhardt P, Ludwig WD, Vandamme AM, Pauli G. Sequence analysis of the first HTLV-I infection in Germany without relations to endemic areas. AIDS Res Hum Retrovir. 1998;14:1199–203.
Schulz TF, Calabro ML, Hoad JG, Carrington CV, Matutes E, Catovsky D, Weiss RA. HTLV-1 envelope sequences from Brazil, the Caribbean, and Romania: clustering of sequences according to geographic origin and variability in an antibody epitope. Virology. 1991;184:483–91.
Miura T, Yamashita M, Zaninovic V, Cartier L, Takehisa J, Igarashi T, Ido E, Fujiyoshi T, Sonoda S, Tajima K, Hayami M. Molecular phylogeny of human T-cell leukemia virus type I and II of Amerindians in Colombia and Chile. J Mol Evol. 1997;44(Suppl 1):S76–82.
Andonov A, Coulthart MB, Perez-Losada M, Crandall KA, Posada D, Padmore R, Giulivi A, Oger JJ, Peters AA, Dekaban GA. Insights into origins of Human T-cell Lymphotropic Virus Type 1 based on new strains from aboriginal people of Canada. Infect Genet Evol. 2012;12:1822–30.
Eirin ME, Berini CA, Jones LR, Dilernia DA, Puca AA, Biglione MM. Stable human T-cell lymphotropic virus type 1 (HTLV-1) subtype a/subgroup a endemicity in Amerindians from Northwest Argentina: a health problem to be resolved. J Med Virol. 2010;82:2116–22.
Eirin ME, Dilernia DA, Berini CA, Jones LR, Pando MA, Biglione MM. Divergent strains of human T-lymphotropic virus type 1 (HTLV-1) within the Cosmopolitan subtype in Argentina. AIDS Res Hum Retrovir. 2008;24:1237–44.
Gascoyne RD, Kim SM, Oger JJ, Melosky BL, Dekaban GA. HTLV-I associated adult T cell leukemia/lymphoma: report of two cases from an Amerindian population in coastal northwest British Columbia. Leukemia. 1996;10:552–7.
Paiva A, Casseb J. Origin and prevalence of human T-lymphotropic virus type 1 (HTLV-1) and type 2 (HTLV-2) among indigenous populations in the Americas. Rev Inst Med Trop Sao Paulo. 2015;57:1–13.
Coulthart MB, Posada D, Crandall KA, Dekaban GA. On the phylogenetic placement of human T cell leukemia virus type 1 sequences associated with an Andean mummy. Infect Genet Evol. 2006;6:91–6.
Gastaldello R, Iniguez AM, Otsuki K, Lamas G, Balangero M, Barbas MG, Mangano A, Sen L, Maturano E, Remondegui C, et al. HTLV type 1 genetic types among native descendants in Argentina. AIDS Res Hum Retrovir. 2008;24:1139–46.
Gessain A, Pecon-Slattery J, Meertens L, Mahieux R. Origins of HTLV-1 in South America. Nat Med. 2000;6:232 (author reply 233).
Azarpazhooh MR, Hasanpour K, Ghanbari M, Rezaee SA, Mashkani B, Hedayati-Moghaddam MR, Valizadeh N, Farid Hosseini R, Foroghipoor M, Soltanifar A, et al. Human T-lymphotropic virus type 1 prevalence in Northeastern Iran, Sabzevar: an epidemiologic-based study and phylogenetic analysis. AIDS Res Hum Retrovir. 2012;28(9):1095–101.
Voevodin A, Gessain A. Common origin of human T-lymphotropic virus type-I from Iran, Kuwait, Israel, and La Reunion Island. J Med Virol. 1997;52:77–82.
Voevodin A, Al-Mufti S, Farah S, Khan R, Miura T. Molecular characterization of human T-lymphotropic virus, type 1 (HTLV-1) found in Kuwait: close similarity with HTLV-1 isolates originating from Mashhad, Iran. AIDS Res Hum Retrovir. 1995;11:1255–9.
Ohkura S, Yamashita M, Ishida T, Babu PG, Koyanagi Y, Yamamoto N, Miura T, Hayami M. Phylogenetic heterogeneity of new HTLV type 1 isolates from southern India in subgroup A. AIDS Res Hum Retrovir. 2005;21:325–30.
Satake M, Yamaguchi K, Tadokoro K. Current prevalence of HTLV-1 in Japan as determined by screening of blood donors. J Med Virol. 2012;84:327–35.
Blattner WA, Nomura A, Clark JW, Ho GY, Nakao Y, Gallo R, Robert-Guroff M. Modes of transmission and evidence for viral latency from studies of human T-cell lymphotrophic virus type I in Japanese migrant populations in Hawaii. Proc Natl Acad Sci USA. 1986;83:4895–8.
Englebrecht S, van Rensburg EJ, Robson BA. Sequence variation and subtyping of human and simian T-cell lymphotropic virus type I strains from South Africa. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;12:298–302.
Mahieux R, de The G, Gessain A. The tax mutation at nucleotide 7959 of human T-cell leukemia virus type 1 (HTLV-1) is not associated with tropical spastic paraparesis/HTLV-1-associated myelopathy but is linked to the cosmopolitan molecular genotype. J Virol. 1995;69:5925–7.
Saito M. Association between HTLV-1 genotypes and risk of HAM/TSP. Front Microbiol. 2019;10:1101.
Einsiedel L, Fernandes L, Spelman T, Steinfort D, Gotuzzo E. Bronchiectasis is associated with human T-lymphotropic virus 1 infection in an Indigenous Australian population. Clin Infect Dis. 2012;54:43–50.
Einsiedel L, Pham H, Wilson K, Walley R, Turpin J, Bangham C, Gessain A, Woodman RJ. Human T-lymphotropic virus type 1c subtype proviral loads, chronic lung disease and survival in a prospective cohort of Indigenous Australians. PLoS Negl Trop Dis. 2018;12:e0006281.
Einsiedel L, Purcell D, Schinke S, Haynes K, Taylor GP, Martin F. Highlights from the HTLV-1 symposium at the 2017 Australasian HIV and AIDS conference held jointly with the 2017 Australasian sexual health conference, November 2017, Canberra, Australia. J Virus Erad. 2018;4:48–50.
Einsiedel L, Cassar O, Bardy P, Kearney D, Gessain A. Variant human T-cell lymphotropic virus type 1c and adult T-cell leukemia, Australia. Emerg Infect Dis. 2013;19:1639–41.
Kirkland MA, Frasca J, Bastian I. Adult T-cell leukaemia lymphoma in an aborigine. Aust N Z J Med. 1991;21:739–41.
Desdouits M, Cassar O, Maisonobe T, Desrames A, Aouba A, Hermine O, Mikol J, Polivka M, Penisson-Besnier I, Marcorelles P, et al. HTLV-1-associated inflammatory myopathies: low proviral load and moderate inflammation in 13 patients from West Indies and West Africa. J Clin Virol. 2013;57:70–6.
Zanella L, Pina-Araujo II, Morgado MG, Vicente AC. Genome-wide analyses of HTLV-1aD strains from Cape Verde, Africa. Mem Inst Oswaldo Cruz. 2016;111:594–6.
Calattini S, Betsem E, Bassot S, Chevalier SA, Tortevoye P, Njouom R, Mahieux R, Froment A, Gessain A. Multiple retroviral infection by HTLV type 1, 2, 3 and simian foamy virus in a family of Pygmies from Cameroon. Virology. 2011;410:48–55.
Padua E, Rodes B, Perez-Pinar T, Silva AF, Jimenez V, Ferreira F, Toro C. Molecular characterization of human T cell leukemia virus type 1 subtypes in a group of infected individuals diagnosed in Portugal and Spain. AIDS Res Hum Retrovir. 2011;27:317–22.
Kjerulff B, Honge BL, Olesen JS, Jensen MM, da Silva ZJ, Erikstrup C, Christiansen M. Phylogeny of human T-lymphotropic virus-1 subtypes in Guinea-Bissau. Trans R Soc Trop Med Hyg. 2018;112:175–80.
Dube DK, Dube S, Erensoy S, Jones B, Bryz-Gornia V, Spicer T, Love J, Saksena N, Lechat MF, Shrager DI, et al. Serological and nucleic acid analyses for HIV and HTLV infection on archival human plasma samples from Zaire. Virology. 1994;202:379–89.
Zanella L, Otsuki K, Marin MA, Bendet I, Vicente AC. Complete genome sequence of Central Africa human T-cell lymphotropic virus subtype 1b. J Virol. 2012;86:12451.
Vicente AC, Gudo ES, Iniguez AM, Otsuki K, Bhatt N, Abreu CM, Vubil A, Bila D, Ferreira OC, Tanuri A, Jani IV. Genetic characterization of human T-cell lymphotropic virus type 1 in Mozambique: transcontinental lineages drive the HTLV-1 endemic. PLoS Negl Trop Dis. 2011;5:e1038.
Etenna SL, Caron M, Besson G, Makuwa M, Gessain A, Mahe A, Kazanji M. New insights into prevalence, genetic diversity, and proviral load of human T-cell leukemia virus types 1 and 2 in pregnant women in Gabon in equatorial central Africa. J Clin Microbiol. 2008;46:3607–14.
Vermeulen M, Sykes W, Coleman C, Custer B, Jacobs G, Jaza J, Kaidarova Z, Hlela C, Gessain A, Cassar O, et al. The prevalence of human T-lymphotropic virus type 1 & 2 (HTLV-1/2) in South African blood donors. Vox Sang. 2019;114:451–8.
Chou KS, Okayama A, Tachibana N, Lee TH, Essex M. Nucleotide sequence analysis of a full-length human T-cell leukemia virus type I from adult T-cell leukemia cells: a prematurely terminated PX open reading frame II. Int J Cancer. 1995;60:701–6.
Ratner L, Philpott T, Trowbridge DB. Nucleotide sequence analysis of isolates of human T-lymphotropic virus type 1 of diverse geographical origins. AIDS Res Hum Retrovir. 1991;7:923–41.
Enose-Akahata Y, Caruso B, Haner B, Charlip E, Nair G, Massoud R, Billioux BJ, Ohayon J, Switzer WM, Jacobson S. Development of neurologic diseases in a patient with primate T lymphotropic virus type 1 (PTLV-1). Retrovirology. 2016;13:56.
Acknowledgements
Not applicable.
Funding
This work received the funding from the “Investissement d’Avenir” as a part of the French Research Program “Laboratoire d’Excellence” (LaBex): Integrative Biology of Emerging Infectious diseases (ANR10-LBX-62 IBEID).
Author information
Authors and Affiliations
Contributions
PVA, OC and AG wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, funless otherwise stated in a credit line to the data.
About this article

Cite this article
Afonso, P.V., Cassar, O. & Gessain, A. Molecular epidemiology, genetic variability and evolution of HTLV-1 with special emphasis on African genotypes. Retrovirology 16, 39 (2019). https://doi.org/10.1186/s12977-019-0504-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-019-0504-z